

More:
News & Stories
Preventing Future Fatalities: Genetic Heart Disease Computationally Modelled
The model of the dysfunctional protein RyR2, developed by Dr. Yulia Einav and her collaborators, may serve as a basis for a quick and effective diagnostic tool for a wide range of life-threatening genetic disorders.
The paper was published in the prestigious Journal of the American Heart Association.

Dr. Yulia Einav
In January 2017 Dr. Yulia Einav of the Bioengineering Department at HIT's Faculty of Engineering received a call that set her mind racing… Prof. David Luria, a renowned electro-cardiologist from the Hadassah Medical Center in Jerusalem, asked if she could help his team unravel the mysteries of a newly discovered mutation. This mutation, he said, had recently caused sudden cardiac arrest in four children of the same family in a village near Jerusalem – killing one excited young girl in the middle of a fun Ferris wheel ride… Dr. Einav, whose own brother suffers from a congenital heart defect, was more than thrilled by the new challenge.
Prof. Luria (who, as Dr. Einav later discovered, had operated twice on her brother, saving his life…) told Dr. Einav of his own findings: the cause of abnormal heart rhythm and resulting cardiac arrests in these children was a mutation in a gene called RyR2, coding for the protein of a channel that delivers calcium (Ca) ions essential for contraction of the heart muscle. When this protein is defective, the channel does not function properly, and heart arrest can occur at any moment – especially at times of excitement, sudden alarm, etc. Mutations in the RyR2 gene and their effects had been known for some time and are estimated to cause 10% of all unexplained sudden cardiac deaths in young people. However, this specific case was different from other RyR2 mutations, particularly in its inheritance pattern within the family. To understand it better, help the affected family and prevent fatal incidents in the future, Prof. Luria asked for Dr. Einav's help.
"I specialize in building computational models of protein molecules," explains Dr. Einav. "Using the data from Hadassah I created a model of the RyR2 protein in the sick children, and compared it to the healthy protein, as well as other defective RyR2 proteins from known literature. Ultimately, we made two important discoveries."
First of all, the new RyR2 mutation was found to be recessive, while in all other known cases it had been dominant. "This means that for a child to have this life-threatening condition, he/she must get the mutated gene from both parents," she says. "It also means that when both parents carry the mutation, there is a 25% chance for each of their children to have the condition. This explains why the new mutation is rarer than those known so far, and why it made its appearance in a family that frequently intermarries within itself. Its recessive quality also clarifies why it remained undiscovered for so long: the parents had been unwitting carriers of defective genes but were not personally affected."
Dr. Einav's second important finding was the location of the defect in the protein itself: "Unlike all other known, clinically manifested mutations in the RyR2 protein, this specific mutation appeared at the protein's periphery. While all others are found close to the crucial region in the protein, where the channel seizes the calcium ion and sends it to the heart muscle cell, this particular defect apparently disrupts the channel's functions from quite a distance. This phenomenon, called an allosteric effect, has never before been discovered for this specific calcium ion channel."
Building various computational models of the protein, Dr. Einav and her team examined different situations, with the mutation inherited from either one parent or both. In this way they were able to show that this specific RyR2 mutation was dose-dependent. In other words, when the defect is far from the calcium channel region, a larger dose, namely 2 copies of the mutated gene (from both parents), is needed, and the mutation is recessive. In this it differs from other known RyR2 mutations, which, being centrally located, need only one copy for the disease to develop, making the mutation dominant.
Supported by these findings (as well as the functional studies of the RyR2 channel performed at Calgary University in Canada), the doctors at Hadassah were able to offer their at-risk patients some help. All members of the family in which the mutation had been discovered were screened, and several more individuals with two copies of the faulty gene were found and cautioned. In addition, genetic counseling is on offer, to prevent marriages between two carriers of the gene, and/or enable prenatal diagnosis of the disease.
The researchers believe that the computational model they have built can serve as a basis for developing a much broader clinical diagnostic tool. "We are currently developing an algorithm that can provide physicians with a quick clinical interpretation of any mutation in the RyR2 gene, at the click of a key," says Dr. Einav. "Once a patient is found to carry the mutation, the physician will simply feed the patient's personal data into the system, and give him/her an accurate diagnosis, indicating whether he/she is at risk of sudden cardiac arrest. Moreover, since the calcium channel addressed in this study is similar to many other critical ion channels (delivering ions of calcium, sodium or potassium) throughout the body - with functions varying from muscle contraction to the propagation of nerve impulses and hormone release - we have begun to formulate similar diagnostic tools for other ion channels as well."
Ultimately, the researchers hope that their study will lead to the development of a range of quick and accurate diagnostic tools for a wide range of mutations potentially affecting ion channels and causing a variety of genetic disorders that threaten the lives and/or quality of life of millions around the world.
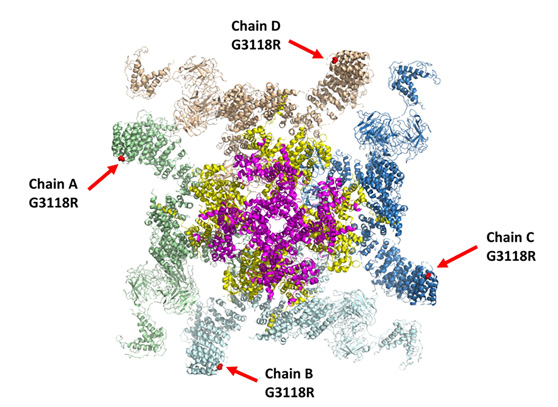
Computational model of RyR2 mutation addressed in the study
The RyR2 calcium channel comprises four protein molecules, shown here in different colors (green, brown, dark blue, light blue). The mutation’s location in each protein is shown as a red dot indicated with a red arrow. Located in a protein’s periphery, this mutation deforms central parts of the channel, marked here with pink and yellow.
Posted: 04/04/2021
- News & Events

New Collaboration with Sheba Medical Center will qualify nurses to work in a digital environment.
Collaboration between HIT Holon Institute of Technology, the teaching authority of the Sheba Medical Center, and the Sheba-BEYOND virtual hospital will allow training nurses in Israel and around the world to work in a digital...